🔬ESMFold2: Bài học cay đắng đang đến với protein - Alex Rives, BioHub
Ghi chú của Biên tập viên: Trong buổi BioHub pod đầu tiên với Priscilla và Mark, họ đã thảo luận về việc mua lại EvoScale, do Alex Rives dẫn dắt, người hiện là Trưởng phòng Khoa học tại BioHub. Với ESM-1, họ đã huấn luyện các mô hình ngôn ngữ trên hàng triệu chuỗi protein được lấy từ khắp các dạng sống, với một mục tiêu "mã thông báo tiếp theo" đơn giản: dự đoán các axit amin đã bị che ngẫu nhiên, dựa trên ngữ cảnh của phần còn lại của chuỗi. Nhưng họ nhanh chóng phát hiện ra rằng các mô hình này cũng học được cấu trúc và chức năng sinh học, bao gồm các thuộc tính mà mô hình chưa bao giờ được hiển thị rõ ràng VÀ khả năng này
Ghi chú của biên tập viên: Trong buổi thảo luận BioHub đầu tiên của chúng tôi với Priscilla và Mark, họ đã thảo luận về việc mua lại EvoScale, do Alex Rives dẫn đầu, người hiện là Trưởng phòng Khoa học tại BioHub. Với ESM-1, họ đã huấn luyện các mô hình ngôn ngữ trên hàng triệu trình tự protein được lấy từ khắp các dạng sống, với mục tiêu "mã thông báo tiếp theo" đơn giản: dự đoán các axit amin đã được che ngẫu nhiên, dựa trên ngữ cảnh của phần còn lại của trình tự. Nhưng họ nhanh chóng nhận thấy rằng các mô hình này cũng học được cấu trúc và chức năng sinh học, bao gồm các thuộc tính mà mô hình chưa bao giờ được hiển thị rõ ràng VÀ khả năng này mở rộng một cách có thể dự đoán được với điện toán, dẫn đến ESM2 và ESM3.
Hôm nay, Alex đã công bố ESMFold 2, một công cụ khoa học mở để thúc đẩy dự đoán, thiết kế và khám phá trong sinh học protein.
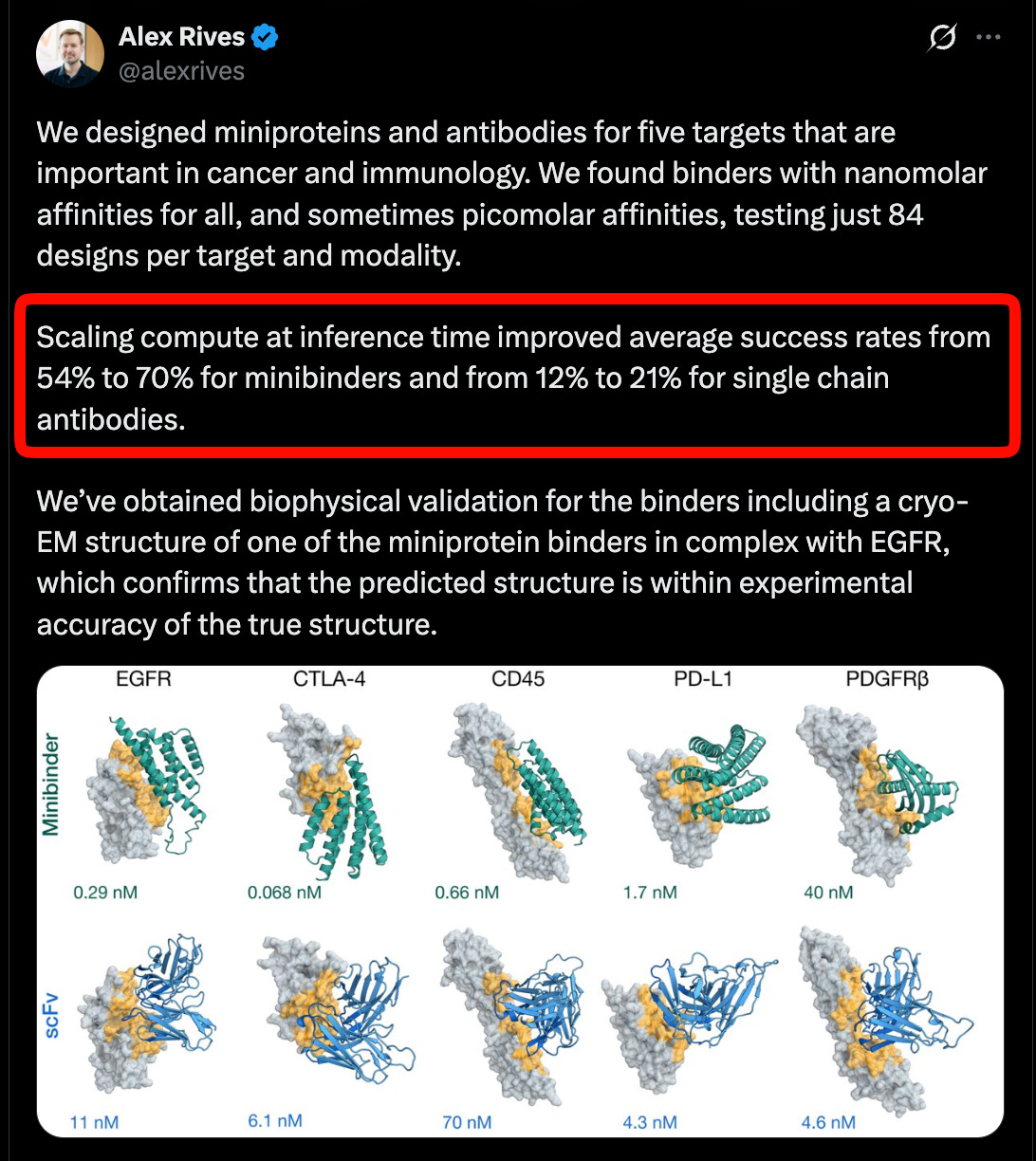
Dựa trên dữ liệu Cryo-EM (đã thảo luận trong buổi thảo luận CZI), ESMFold2 báo cáo hiệu suất tiên tiến về tương tác protein, đặc biệt là kháng thể, một phương thức quan trọng cho liệu pháp, và bằng chứng cho thấy việc mở rộng thời gian suy luận cũng hoạt động trên năm mục tiêu trong ung thư và miễn dịch học.
Để ghi nhận dự án AI x gấp protein nổi tiếng khác, họ cũng đang phát hành một tập hợp 6,8 tỷ protein và 1,1 tỷ cấu trúc được dự đoán, mà bạn có thể thử nghiệm trên trang web của họ. Chúng tôi rất vinh dự được hợp tác với họ trong lần phát hành lớn này!
Một trong những điệp khúc chúng tôi đã nghe trong buổi thảo luận Khoa học là gấp protein, thiết kế vật liệu, sinh học tế bào, v.v. là những vấn đề rất khác so với Mô hình Ngôn ngữ. Chúng chắc chắn là như vậy. Tuy nhiên, Alex Rives và nhóm ESM tại BioHub vừa phát hành một bản in trước và mô hình, chứng minh rằng các mô hình biến đổi kiểu BERT thông thường được huấn luyện trên các tập dữ liệu đủ lớn và đa dạng có thể đánh bại các mô hình chuyên biệt như AlphaFold3 trên một số vấn đề khó nhất liên quan đến protein.
Andrew White đã có một phần tuyệt vời trong tập LS-Science đầu tiên của chúng tôi, giải thích AlphaFold2 đã gây kinh ngạc như thế nào khi nó được phát hành vào năm 2020: nó đột nhiên giải quyết các vấn đề trên GPU trên máy tính để bàn của bạn mà DESRes đã xây dựng các cụm siêu máy tính ASIC tùy chỉnh để giải quyết. John Jumper và Demmis Hassabis đã nhận giải Nobel Hóa học cho công trình này.
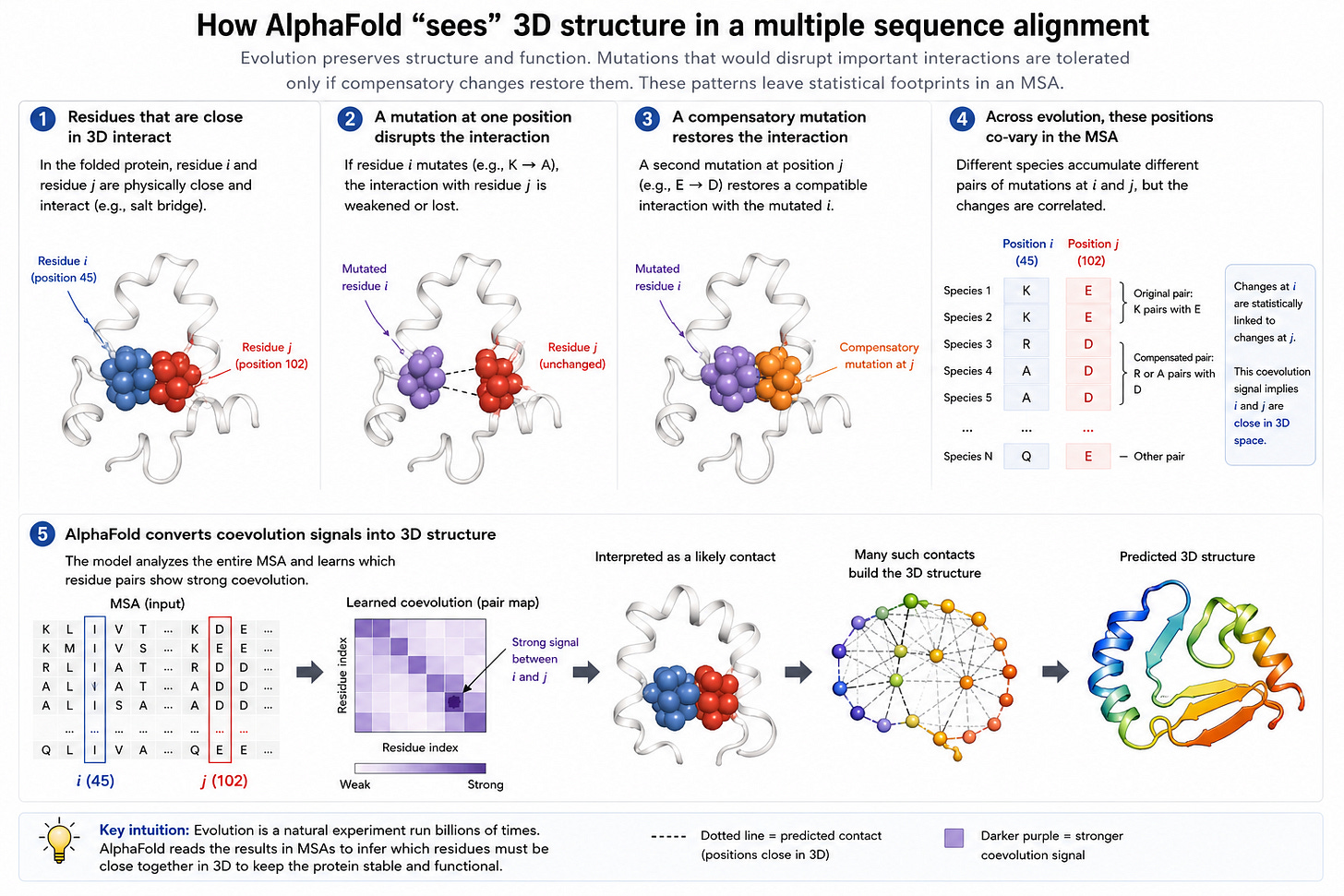
AlphaFold2 đã tận dụng một quan sát rất thông minh: nếu nhiều loài cùng tiến hóa các cặp đột biến, điều này ngụ ý rằng các đột biến tương ứng với các phần của protein gần nhau trong không gian 3D. Điều này thường được viết tắt là MSA (multi-sequence alignments - căn chỉnh đa trình tự), và là hiểu biết quan trọng giúp AlphaFold2 rất hiệu quả.
Tuy nhiên, giống như các thiên kiến quy nạp khác, nó làm tổn hại đến khả năng khái quát hóa.
Đã "nghiện" mở rộng quy mô trước khi nó trở nên phổ biến
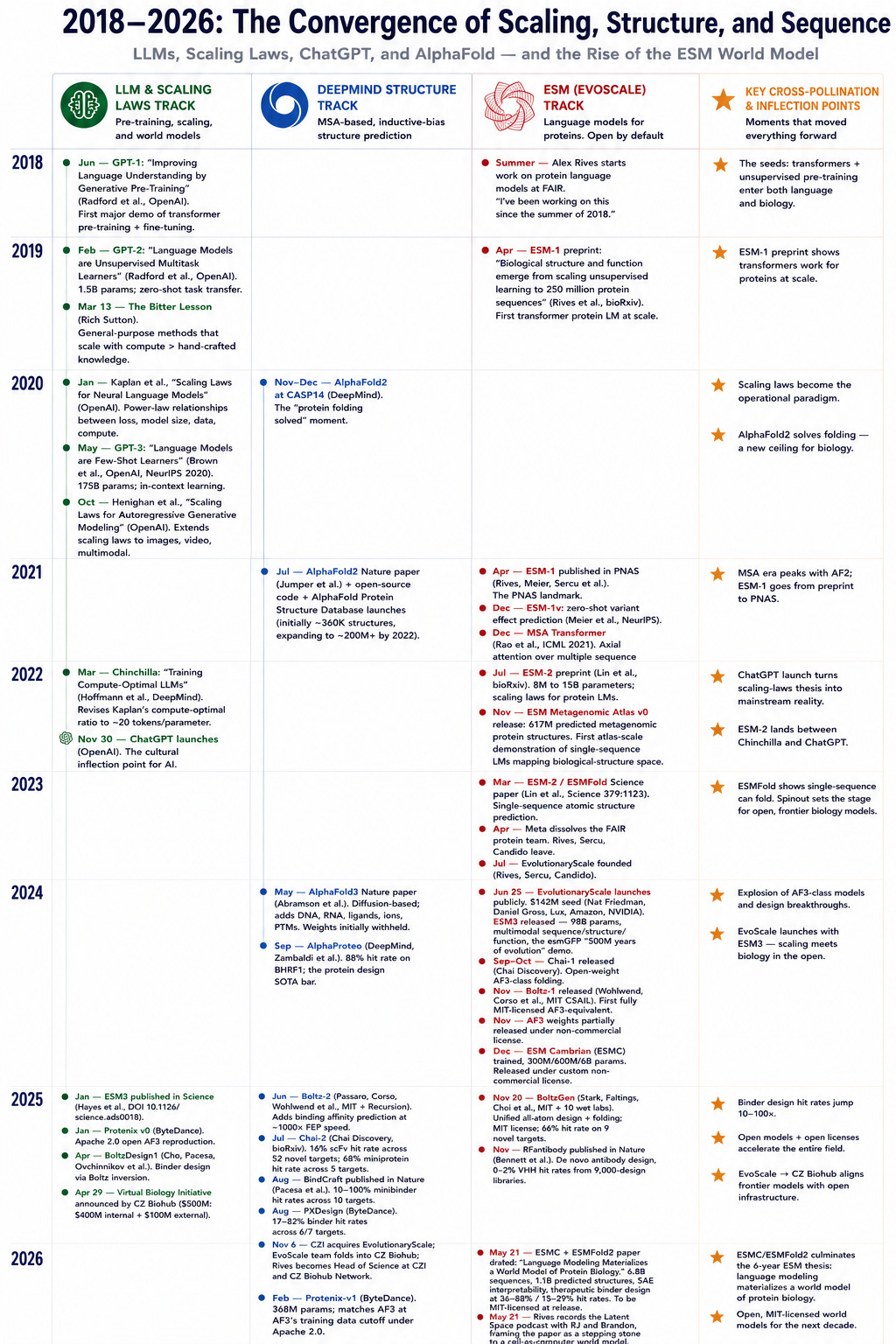
Nếu bạn xem xét dòng thời gian cho các quy luật mở rộng quy mô đối với LLM và việc phát hành các mô hình dự đoán cấu trúc1, nhóm ESM đáng chú ý đã tăng cường cách tiếp cận "bất chấp MSA" của họ sau khi AlphaFold2 được phát hành. Điều này rõ ràng đòi hỏi một niềm tin lớn vào giả thuyết mở rộng quy mô.
Tại sao lại có niềm tin đó?
ESM được phát triển vào thời điểm nhiều quy luật mở rộng quy mô và "Bài học cay đắng" ngày càng được chứng minh là đúng. Thành công vang dội của AlphaFold2 chắc hẳn vừa thú vị vừa gây thất vọng sâu sắc. Nhưng việc sử dụng MSA có nghĩa là mô hình phụ thuộc vào dữ liệu huấn luyện có chứa MSA để chính xác trong một miền nhất định. Đối với những thứ như kháng thể không có MSA để huấn luyện2, AlphaFold có xu hướng hoạt động kém.
ESM tiếp cận theo một cách khác: tìm hiểu mối quan hệ giữa các protein khác nhau bằng cách huấn luyện không giám sát trên càng nhiều dữ liệu đa dạng càng tốt (nghe có quen không?) và sau đó đối chiếu lại với các cấu trúc đã biết từ Ngân hàng Dữ liệu Protein (PDB) và các nguồn khác.
Nói cách khác, đây là một Mô hình Thế giới.
Mô hình Thế giới cho protein
"Mô hình Thế giới" là một thuật ngữ cường điệu mà tôi định nghĩa như sau:
Sử dụng huấn luyện không giám sát để học các mẫu trừu tượng từ dữ liệu:
Sự trừu tượng phải mang tính ngữ nghĩa – các cấu trúc mới đại diện cho những thứ tuân theo quy tắc của thế giới thực.
Sự trừu tượng phải mang tính cấu trúc – việc kết hợp lại các mẫu khác nhau dẫn đến các cấu trúc mới và thường hợp lệ.
Sự trừu tượng phải hỗ trợ khái quát hóa – nó dự đoán những điều trong thế giới thực mà nó chưa được huấn luyện.
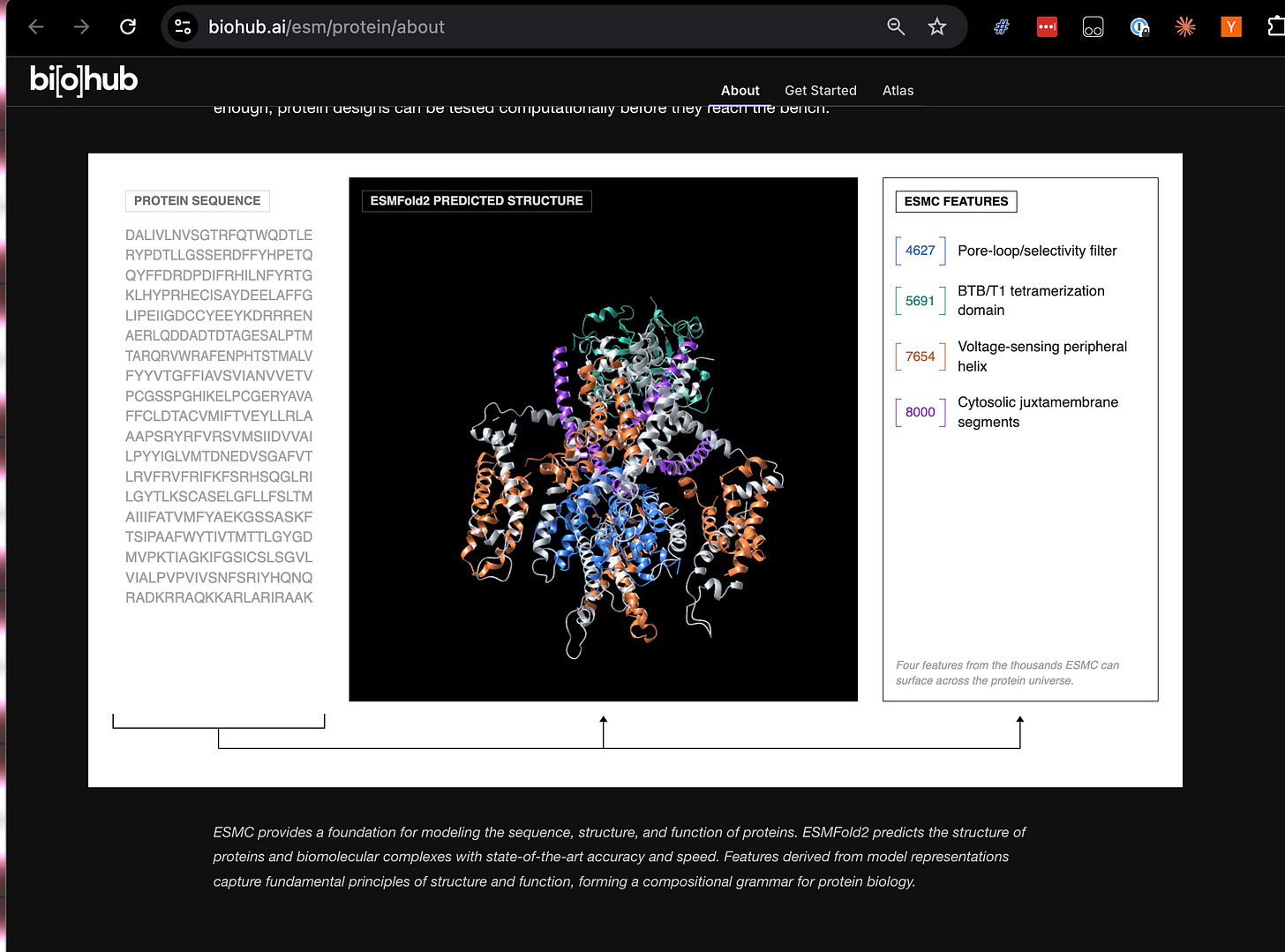
Khi có một mô hình thế giới, có thể gắn thêm các "đầu" vào đó cho các tác vụ tiếp theo: dự đoán các đặc tính của protein, phân tích các đặc điểm chức năng của nó, hoặc tìm kiếm trong biểu diễn các protein đáp ứng tiêu chí thiết kế. Hai mô hình lớn mà BioHub vừa phát hành theo giấy phép MIT trực tiếp phù hợp với điều này:
Mô hình thế giới → ESMC (một mô hình được huấn luyện trên 2,8 tỷ chuỗi)
Đầu dự đoán cấu trúc → ESMFold2
Một trong những cách thú vị mà mô hình thế giới có thể "dự đoán mọi thứ" là tạo ra các chuỗi protein và sau đó đo lường các đặc tính dự đoán, chẳng hạn như ái lực liên kết, trong phòng thí nghiệm. Alex đã nói trong tập về việc xác nhận một số phân tử khó hơn mà họ đã dự đoán trong phòng thí nghiệm. Rất tuyệt vời!
Một cách khác là sử dụng các kỹ thuật giải thích cơ chế như Bộ mã hóa tự động thưa (SAE) để trích xuất các đặc điểm ngữ nghĩa từ mô hình, sau đó tìm các đặc điểm mới dự đoán sinh học chưa biết. Tôi sẽ không tiết lộ phần này cho bạn: đó là một trong những điểm nổi bật của tập đối với tôi!
Một tế bào là một máy tính
Tất cả chúng ta đều đã nghe nói rằng gen giống như các chương trình máy tính, nhưng thường thì sự tương tự đó chỉ dừng lại ở đó. Tất nhiên, gen được phiên mã thành RNA và RNA được dịch mã thành protein, vì vậy gen là chương trình để xây dựng protein, nhưng điều đó chỉ mang sự tương tự đến mức "các bit nhị phân là chương trình".
Đây là một sự tương tự tốt hơn: có thể coi nhân tế bào là một thiết bị lưu trữ/bộ điều khiển lưu trữ, ribosome là một trình biên dịch và thời gian chạy JIT, và các đặc điểm ngữ nghĩa mà chúng ta học được từ mô hình thế giới của mình thông qua SAE là các hàm, protein là các quá trình tương tác với nhau trong các quy trình làm việc (đường dẫn tín hiệu) để tạo ra các hành vi và đầu ra (kiểu hình).
Giống như các hàm, các đặc điểm SAE có cấu trúc phân cấp từ cấu trúc cục bộ, thứ cấp và bậc ba (mô phỏng cấu trúc protein), nhưng cũng có các motif mang tính khái niệm.
Nguồn tin: Latent Space — Tác giả: RJ Honicky. Bản dịch tiếng Việt do AI thực hiện, có thể có sai sót.